Week 6: Project 1 And Midterm 1
CONTENTS:
- Learning Goals, Readings, Background, and PreQuiz
- Midterm Exam 1
- Recitation
- Recitation Submission Guidelines
- Project 1
- Introduction
- g++ flags
- Assignment
- Question 1: isValidBase()
- Question 2: isValidStrand()
- Question 3: strandSimilarity()
- Question 4: bestStrandMatch()
- Question 5: Mutation Detection
- Question 6: DNA Sequence Transformations
- Question 7: Reverse Complement of a DNA Sequence
- Question 8: Extracting Reading Frames
- Question 9: Tying It All Together
Learning Goals, Readings, Background, and PreQuiz
Due to the upcoming midterm and project 1, many sections are removed from this weeks workbook. Take this opportunity to go over previous modules 📚 and get upto speed, so you can perform your best on the exam 💪
Midterm Exam 1
Your Exam is Wednesday February 19. See Canvas for more details
Recitation
Recitation Spot The Error - Problem 1
The code snippet below is intended to print the numbers from 1 through 10. Identify the error(s) in the code below, and write the correct line(s).
#include <iostream>
using namespace std;
int main() {
int num = 1;
while(num <= 10) {
cout << num << " ";
int num = num + 1;
}
return 0;
}
Recitation Spot The Error - Problem 2
The code snippet below is supposed to print even numbers less than 9. Identify the error(s) in the code below, and write the correct line(s).
#include <iostream>
using namespace std;
int main() {
int even = 2;
while (even != 9) {
cout << even << " ";
even += 2;
}
return 0;
}
Recitation Pseudocode Analysis - Problem 3
What output does the following pseudocode produce?
Start
Declare an integer variable `m` and initialize it to 0.
Declare an integer variable `i` and initialize it to 1.
Do:
Declare an integer variable `s` and initialize it to 1.
If `i` is not equal to 1 and the remainder of `i` divided by 2 equals 0, then:
Multiply `s` by -1 and assign the result back to `s`.
Print the product of `m` and `s`.
Else:
Increment `m` by 3.
Print the value of `m`.
Increment `i` by one.
While `i` is less than or equal to 5.
End
Recitation Tax Season! – Problem 4
Note: The following scenario is entirely fictional and any resemblance to real life situations is purely coincidental. Provided numbers should not be interpreted as financial advice. CSCI 1300 is not liable for any misinterpretation of this information. 🤣
As spring semester unfolds at CU Boulder, tax season arrives for students who’ve had part-time jobs, internships, or side hustles. Many are filing taxes for the first time and want to get it right (and maybe even get a refund!). Your task: create a C++ tax calculator that applies a standard deduction, an optional dependent deduction, and then selects the correct tax bracket based on the remaining taxable income.
- Standard Deduction: $14,600 (applies to everyone)
- Dependent Deduction: $500 additional deduction if you have at least one dependent
After deductions are applied, the taxable income is determined and placed into one of three brackets:
- Bracket A: Taxable income ≤ $10,000 ⇒ taxed at 10%
- Bracket B: Taxable income > $10,000 and ≤ $50,000 ⇒ taxed at 15%
- Bracket C: Taxable income > $50,000 ⇒ taxed at 20%
Your net income is the original gross income minus the total tax owed.
/*
@brief Applies standard and dependent deductions.
@param income The gross income before deductions.
@param numDependents The number of dependents for the taxpayer.
@return double - the taxable income after all deductions have been applied.
*/
double applyDeductions(double income, int numDependents) {
// TODO: Fill in your logic
}
/*
@brief Calculates the tax owed based on the taxpayer's taxable income.
@param taxableIncome The income amount after deductions have been applied.
@return double - the tax owed.
*/
double calculateTax(double taxableIncome) {
// TODO: Fill in your logic
}
/*
HINT: call calculateTax and applyDeductions from inside of this function.
@brief Computes the net income (gross - tax).
@param grossIncome The original, pre-deduction income.
@param numDependents The number of dependents for the taxpayer.
@return double - the net income after taxes and deductions.
*/
double computeNetIncome(double grossIncome, int numDependents) {
// TODO: Fill in your logic
}
Recitation Tax Season! - Problem 4.a.: Algorithm
Write out the steps you would use to solve this problem by hand as pseudocode.
Recitation Tax Season! - Problem 4.b.: Examples
Pick two possible inputs for each of your three functions (six total). Follow the steps you wrote for these values to find your result, and verify it.
Recitation Tax Season! - Problem 4.c.: Assert Statements
Translate your inputs and expected outputs into assert statements.
Recitation Tax Season! - Problem 4.d.: Implementation
Translate your pseudocode into a C++ program to solve the above code, using your assert statements in your main function to verify that your program works as expected.
Recitation Submission Guidelines
Important: Follow these instructions carefully when preparing your recitation assignments. Your final submission should be in a single document, and the only action required on Canvas is uploading that document.
- Documentation:
- Create a pdf that includes your submission for all recitation questions. This is the pdf you will upload to your canvas assignment. Feel free to use Word/Google doc to create the pdf.
- Clearly label each question with its corresponding number and include content as applicable (see #2).
- Content to Include:
- Screenshots of Your Code:
- For each question, include a screenshot of your code.(corrected code in case of spot the errors)
- Screenshots of Code Output (if applicable):
- For some longer questions, it might be required to take a screenshot of the code’s output. Include these screenshots as part of your submission.
- Longer Recitation Questions (Multiple Parts):
- Option A:
- Comment your answers directly within your code file.(Spot the errors)
- Take screenshots of the commented code and paste them into your document.
- Option B:
- Take screenshots of the unmodified code.
- Write your answers (Free Response/Pseudocode/Edge case identifictation) to the subquestions in the pdf document next to the corresponding screenshots.
- Option A:
- Screenshots of Your Code:
- Submission:
- Upload the final pdf document to Canvas. This is the only action required on Canvas for your submission.
By following these steps, your submission will be clear, organized, and standardized across all recitation assignments.
Project 1
The project is due Friday February 28. We strongly recommend that you pace yourself on the project and start early. You do not have to start the project prior to the midterm exam, however some students find that working on the project helps to improve their exam performance as it gives them more practice with the material.
Introduction
You have recently been employed by Rhonda Labs, a leading research institute working to uncover the secrets of rare genetic diseases. Your first task is to write a program that analyzes DNA from various species and individuals, compares their similarities, and performs analyses on the given sequences.
Before you dive into the project, let’s review a bit of biology to help you understand what you will be working with. DNA, or deoxyribonucleic acid, is the molecule that carries genetic information in almost all living organisms. It acts like a set of instructions that tells cells how to function, grow, and reproduce.
DNA is made up of four chemical bases:
1. Adenine (A)
2. Cytosine (C)
3. Guanine (G)
4. Thymine (T)
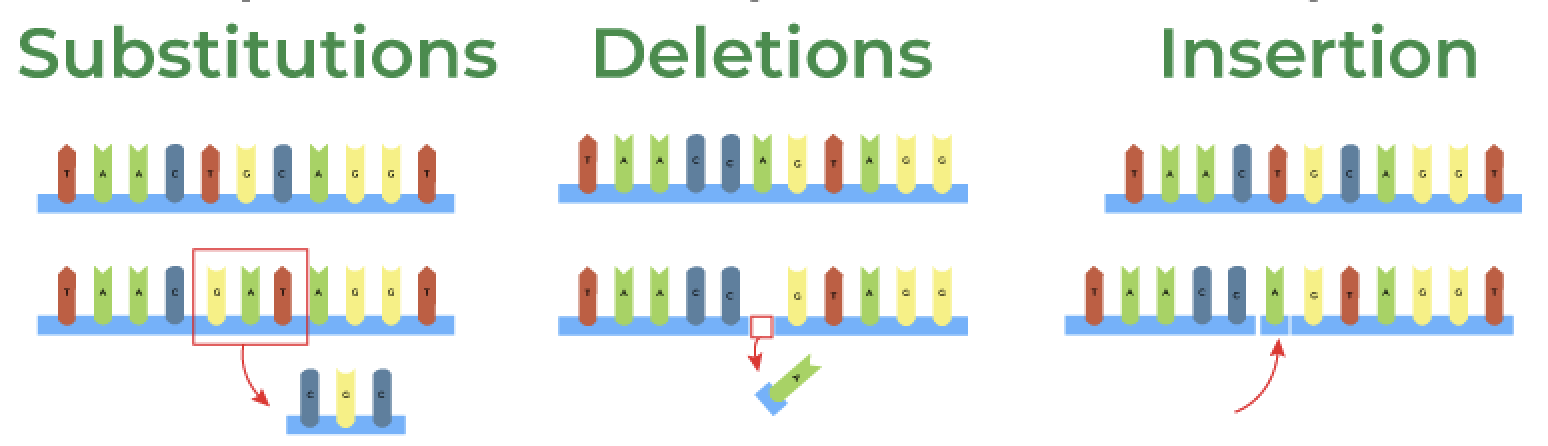
These bases pair together (A with T, C with G) to form the DNA double helix, which is the the overall structure of DNA. A DNA strand is made up of a sequence of these base pairs. Every organism has its own unique DNA sequence, though many organisms share similarities, especially related species. Sometimes, DNA sequences can change, leading to mutations, such as:
1. Substitutions (one base is swapped for another)
2. Insertions (extra bases are added)
3. Deletions (bases are removed)
For this task, the steps are outlined and divided into individual questions. By the end of the project, you will have developed a cohesive program that a user can interact with by inputting their DNA sequences. Below is a breakdown of the steps:
1. Check whether your DNA strands are composed of valid bases
2. Develop functions to compare DNA strands and assess their similarity
3. Develop a function that compares two DNA sequences and identifies all types of mutations between them
4. Transcribe DNA to RNA and compute the reverse complement of a DNA strand
5. Find open reading frames within a DNA strand
6. Final step: make it more accessible and user-friendly!
g++ flags
A point of particular frustration for many, to ensure that your local builds look like our autograder be sure to build using the following flags. Moving forward in the course, we expect this as your standard build procedure.
g++ -Wall -Werror -Wpedantic -Wsign-compare -std=c++17 myProjectFile.cpp
Assignment
Warning: You are not allowed to use global variables for this project.
All function names, return types, and parameters must precisely match those shown. You may not use pass by reference or otherwise modify the function prototypes. You are welcome to create additional functions that may help streamline your code. You must well comment your code, and follow good formatting practices. You should create assert statements for each function to test your code in VS Code before moving to CodeRunner.
Question 1: isValidBase()
One of the first steps when working with DNA sequences is to check whether the data is valid or corrupted. You must ensure the sequences you are working with consist only of valid DNA bases: A, C, G, and T. You will write a function, isValidBase(), that checks if a character is a valid DNA base.
| Function: isValidBase(char) | bool isValidBase(char base) |
| Purpose: | The function will determine whether a given character is a valid DNA base. The function should not print anything. |
| Parameters: | char base - The character to validate |
| Return Value: | The function should return true if the character is a valid base (A, C, G, or T) and false otherwise. |
| Error handling/ Boundary conditions: | - The function should be case sensitive, e.g., ‘A’ is a valid base, but ‘a’ is not. - Note: True is represented by 1 and false by 0 when you cout boolean variables. |
For Question 1, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste only the isValidBase() to the answer box!
| Function call | Expected return value |
| isValidBase(‘A’) | true |
| isValidBase(‘X’) | false |
| isValidBase(‘a’) | false |
| isValidBase(‘T’) | true |
Question 2: isValidStrand()
Next, write the isValidStrand() function that checks if a string contains only valid DNA bases.
| Function: isValidStrand(string) | bool isValidStrand(string strand) |
| Purpose: | The function will determine whether a given string consists only of valid DNA bases. The function should not print anything. |
| Parameters: | string strand - The DNA strand to validate. |
| Return Value: | The function should return true if the string is a valid DNA strand and false otherwise. |
| Error handling/ Boundary conditions: | - The input string is only considered valid if it consists only of A, C, T, and G bases. - If the string is empty, then it should not be considered a valid DNA strand, and your function should return false. - Hint: The function should make use of your isValidBase function. |
For Question 2, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste the isValidBase() and isValidStrand() to the answer box!
| Function call | Expected return value |
| isValidStrand(“ATGCTTCAA”) | true |
| isValidStrand(“CTTZ”) | false |
| isValidStrand(“”) | false |
| isValidStrand(“ATCG”) | true |
Question 3: strandSimilarity()
Comparing DNA sequences allows researchers to identify similarities and differences that may be significant in understanding genetic relationships or disease mechanisms. In this step, you’ll develop functions to compare DNA strands and assess their similarity.
For two DNA strands of equal length, the similarity score is calculated based on the number of matching bases at corresponding positions using the following formula:
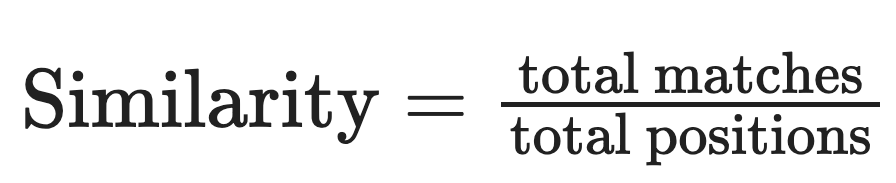
Example: Let’s compare the following two DNA strands:
| Position | Strand 1 | Strand 2 |
| 1 | G | G |
| 2 | A | T |
| 3 | T | T |
| 4 | C | C |
| 5 | A | A |
| 6 | G | A |
The total number of matches is 4 out of 6 positions, resulting in a similarity score of 4/6 = 0.667.
HINT: recall from week 4s reading on “Testing Double Functions” that we need to be careful when compairing doubles. Consider adding and calling the following helper function for your testing:
// This is a required function to successfully test Double functions in C++:
/*
- doublesEqual will test if two doubles are equal to each
- other within two decimal places.
*/
bool doublesEqual(double a, double b, const double epsilon = 1e-2)
{
double c = a - b;
return c < epsilon && -c < epsilon;
}
The function strandSimilarity() compares two strands position by position, counting the number of positions where the bases are identical. This provides a direct measure of how similar the two sequences are.
| Function: strandSimilarity(string, string) | double strandSimilarity(string strand1, string strand2) |
| Purpose: | The function will find the similarity between two DNA strands. The function should not print anything. |
| Parameters: | string strand1 - The first DNA strand to compare. string strand2 - The second DNA strand to compare. |
| Return Value: | The function should return the similarity score between the two strands. |
| Error handling/ Boundary Condition: | - The parameters should be two strings of equal length. If they are not equal in length, your function should return 0. - You may assume that the input to strandSimilarity() will always be a valid strand, i.e., you do not have to account for arbitrary strings. |
HINT: to test the comarisons of doubled use the doublesEqual function described in week 4 of this workbook.
For Question 3, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste the strandSimilarity() and any helper function(s) you used to the answer box!
| Function call | Expected return value |
| strandSimilarity(“AGGT” , “CTGA”) | 0.25 |
| strandSimilarity(“CCTT” , “CCTT”) | 1 |
| strandSimilarity(“ATG” , “AAATTT”) | 0 |
| strandSimilarity(“CTGTAGAGCT” , “TAGCTACCAT”) | 0.2 |
Question 4: bestStrandMatch()
In bestStrandMatch(), the strands can be different lengths, therefore, you’ll want to compare overlapping sections and calculate similarity scores at each position, and to do that you’ll slide the shorter strand along the longer strand. The maximum score across all positions indicates the best alignment between the two strands.
| Function: bestStrandMatch(string, string) | int bestStrandMatch(string input_strand, string target_strand) |
| Purpose: | - The function will find the best similarity between two DNA strands. - The function should print out the best similarity score. |
| Parameters: | string input_strand - The input DNA strand to be checked against the target_strand (length greater than or equal to the target strand) string target_strand - The target DNA strand. |
| Return Value: | If the parameters are valid, returns an int representing the starting index of the substring in the input strand where the best alignment with target strand occurs. |
| Error handling/ Boundary conditions: | - If the input strand is shorter than the target strand, the function returns -1 as the alignment index and prints out “Best similarity score: 0.0”. - This function should make use of the strandSimilarity() function. - You may assume that the input to bestStrandMatch() will always be a valid DNA sequence, i.e., you do not have to account for arbitrary strings. |
HINT: For this problem, lookup the string method named substr. Given a string, a starting position and an length, it will find a smaller “sub-string” of the original. recall that strings index from 0. Some examples below
string x = "hello";
cout << x << endl; // prints "hello"
string y = x.substr( 0, 2 );
cout << y << endl; // prints "he" start at 0, length 2
string z = x.substr( 2, 3 );
cout << z << endl; // prints "llo" start at 2, length 3For Question 4, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste the bestStrandMatch() and any helper function(s) you used to the answer box!
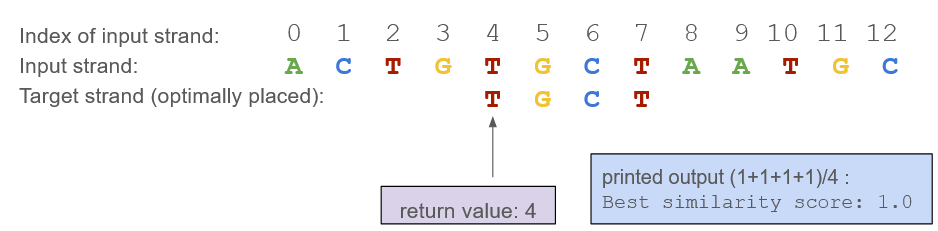
| Function call | Expected return value | Expected printed value |
| bestStrandMatch(“GATCAGT”, “TCA”) | 2 | Best similarity score: 1.0 |
| bestStrandMatch(“AACCTGAC”, “ACT”) | 1 | Best similarity score: 0.666667 |
| bestStrandMatch(“CTG”, “CCCC”) | -1 | Best similarity score: 0.0 |
| bestStrandMatch(“ATCGTA”, “TTCGAT”) | 0 | Best similarity score: 0.5 |
Question 5: Mutation Detection
Now, you want to be able to make deeper observations between DNA sequences. To do this, you will create a function that compares two DNA sequences and identifies all types of mutations between them. The function should align the sequences based on the best possible match and then process the sequences character by character, printing out mutations as they are detected.
Your function should be able to identify the following mutations:
1. Substitution: When bases at the same position differ
2. Insertion: When an extra base is present in the target strand
3. Deletion: When a base from the input strand is missing in the target strand
The function should determine the longest of the two strands and use the bestStrandMatch() function to optimally align them. Upon alignment, the function should print out the best alignment index. After alignment, it should compare the sequences character by character to identify mutations. It detects substitutions when bases at the same aligned position differ, deletions when extra bases are present in the input strand but not in the target strand, and insertions when bases are present in the target strand but missing from the input strand.
| Function: identifyMutations(string, string) |
void identifyMutations(string input_strand, string target_strand) |
| Purpose: | The function compares two DNA sequences to identify all types of mutations between them. It aligns the sequences based on the best possible match and processes them character by character, printing out any mutations as they are detected. |
| Parameters: | string input_strand - The input strand to be checked against the target string target_strand - The target strand |
| Return Value: | N/A |
| Error handling/ Boundary conditions: | -You may assume that the input and target strands are both valid DNA strands. - If no mutations are found, the function outputs “No mutations found.” |
For Question 5, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste identifyMutations() and any helper function(s) to the answer box!
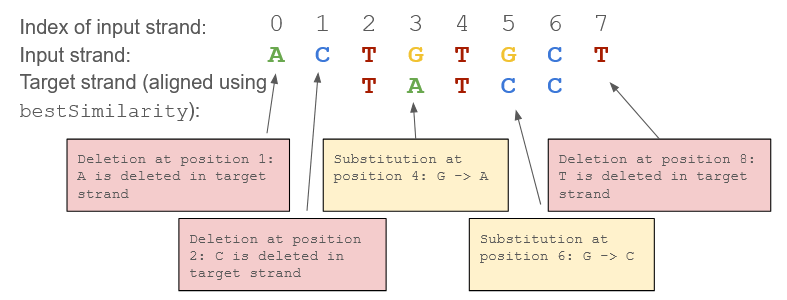
Note: “Best Similarity Score:…” print line should come from your bestStrandMatch() function.
| Function call | Expected printed value |
| identifyMutations(“GGG”, “TAGGGTA”) | Best similarity score: 1 Best alignment index: 2 Insertion at position 1: T is inserted in target strand Insertion at position 2: A is inserted in target strand Insertion at position 6: T is inserted in target strand Insertion at position 7: A is inserted in target strand |
| identifyMutations(“AACCGG”, “AACCGG”) | Best similarity score: 1 Best alignment index: 0 No mutations found. |
| identifyMutations(“TA”, “TAGG”) | Best similarity score: 1 Best alignment index: 0 Insertion at position 3: G is inserted in target strand Insertion at position 4: G is inserted in target strand |
| identifyMutations(“TAGG”, “TA”) | Best similarity score: 1 Best alignment index: 0 Deletion at position 3: G is deleted in target strand Deletion at position 4: G is deleted in target strand |
| identifyMutations(“AGTCACG”, “AGCTACA”) | Best similarity score: 0.571429 Best alignment index: 0 Substitution at position 3: T -> C Substitution at position 4: C -> T Substitution at position 7: G -> A |
Question 6: DNA Sequence Transformations
In this part, you will simulate the transcription process by converting a DNA sequence into an RNA sequence. This involves replacing every occurrence of thymine (‘T’) with uracil (‘U’) in the DNA strand.
| Function: transcribeDNAtoRNA(string) | void transcribeDNAtoRNA(string strand) |
| Purpose: | The function will transcribe a DNA sequence to RNA and print the RNA sequence to the console. The function will replace all occurrences of ‘T’ with ‘U’. |
| Parameters: | string strand - The DNA sequence to be transcribed. |
| Return Value: | N/A |
| Error handling/ Boundary conditions: | You may assume that the input DNA strand is a valid DNA sequence. |
For Question 6, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste transcribeDNAtoRNA() and any helper function(s) to the answer box!
| Function call | Expected printed value |
| transcribeDNAtoRNA(“ATCGTACG”) | AUCGUACG |
| transcribeDNAtoRNA(“TTAA”) | UUAA |
| transcribeDNAtoRNA(“ACCG”) | ACCG |
| transcribeDNAtoRNA(“T”) | U |
Question 7: Reverse Complement of a DNA Sequence
In this part, you will write a function to compute the reverse complement of a DNA strand. The reverse complement is obtained by reversing the DNA sequence and then replacing each base with its complement:
1. 'A' is complemented by 'T'
2. 'T' is complemented by 'A'
3. 'C' is complemented by 'G'
4. 'G' is complemented by 'C'
| Function: reverseComplement(string) | void reverseComplement(string strand) |
| Purpose: | The function will compute the reverse complement for a DNA sequence and print the result to the console. |
| Parameters: | string strand - The DNA sequence for which the reverse complement will be computed. |
| Return Value: | N/A |
| Error handling/ Boundary condition: | You may assume that the input DNA strand is a valid DNA sequence. |
For Question 7, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste the reverseComplement() and any helper function(s) you used to the answer box!
| Function call | Expected printed value |
| reverseComplement(“ATCGTACG”) | CGTACGAT |
| reverseComplement(“AAACCC”) | GGGTTT |
| reverseComplement(“AGCT”) | AGCT |
| reverseComplement(“A”) | T |
Question 8: Extracting Reading Frames
When working with DNA sequences, it is often necessary to focus on specific regions called open reading frames (ORF). These DNA regions are read in groups of three bases, called codons, which are later translated into amino acids during the process of protein synthesis. An ORF is a continuous sequence of DNA that begins with a start codon (ATG) and ends with a stop codon (TAA, TAG, or TGA).
Your task is to identify valid protein-coding regions in a DNA sequence. A valid ORF starts with the ATG codon and ends with one of the stop codons (TAA, TAG, or TGA), with the number of bases between the start and stop codons being divisible by 3.
| Function: getCodingFrames(string) | void getCodingFrames(string strand) |
| Purpose: | The function will print out complete reading frames. |
| Parameters: | string strand - The DNA strand from which to extract reading frames. |
| Return Value: | N/A |
| Error handling/ Boundary condition: | - If no reading frames are found, the function should print “No reading frames found.” - You may assume that the input DNA strand is a valid DNA sequence. - **Note: **There could be multiple ORF within a single DNA strand. |
For Question 8, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste the getCodingFrames() and any helper function(s) you used to the answer box!
| Function call | Expected printed value |
| getCodingFrames(“ATGCGGTAA”) | ATGCGGTAA |
| getCodingFrames(“AAACCC”) | No reading frames found. |
| getCodingFrames(“ATGCGTAGCTAAATGGGGTAG”) | ATGCGTAGCTAA ATGGGGTAG |
Question 9: Tying It All Together
You have successfully written functions to help your research team analyze DNA sequences! However, now you want to make it more accessible and user-friendly. For this question, you will create a main function that allows a user to interact with your program by providing their DNA sequences. Your main function should present the user with a menu containing the following options:
1. Calculate the similarity between two sequences of the same length
2. Calculate the best similarity between two sequences of either equal or unequal length
3. Identify mutations
4. Transcribe DNA to RNA
5. Find the reverse complement of a DNA sequence
6. Extract coding frames
7. Exit
Your menu should run on a loop, continually offering the user each option until they choose to exit. Be sure to use the functions you wrote in questions 1 through 6 as needed.
Note: Your main function should account for any user input that isn’t a valid sequence. If user input is not a valid sequence, your program should print "Invalid input. Please enter a valid sequence." until the user enters a valid sequence. Additionally, functions that require strings of the same length should have their inputs validated for matching lengths before being called in the user menu. If the strands are not of the same size, the program should display "Error: Input strands must be of the same length." and return to the menu.
For Question 9, develop and validate your solution in VS Code. Once you are happy with your solution, go to coderunner on Canvas and paste you entire program to the answer box!